Follow us on Linked
Each beneficiary should budget its own costs. Costs should be budgeted in line with the project activities each partner is involved in.
In the conduct of a clinical trial, a sponsor is an individual, institution, company or organization.
You can register via our website, and after an internal assessment you could be added to our pool of reviewers.
The signature of the accession form is due within 30 days from signature of the grant agreement.
No, consortia are formed by applicants without EDCTP involvement.
No, EDCTP uses a different list of experts, but some may be those listed on the EC Portal.
Funding from WHO can be considered as cofunding (additional funding on top of the EDCTP grant amount).
NID = Neglected Infectious Diseases, otherwise also known as Neglected Tropical Diseases.
The work plan for 2015 is currently being reviewed by the European Commission and EDCTP expects a decision no earlier than September 2015.
It can be considered if it is presented as comorbidity with a poverty-related infectious disease within the remit of the call.
Yes. There should be no big deviations to the budget specified in the first stage. But you can make small corrections.
If you mean the second phase of the SME instrument: then yes. Otherwise: no.
No. The only costs eligible before project start are travel costs to the kick-of-meeting IF the travel itself took part within the project duration.
Yes. You have to depreciate according to the rules applicable for your company.
Since the price is charged by a different department of the same legal entity, this is considered internal invoicing.
No, contracting between beneficiaries is not allowed. Each beneficiary must declare its own costs.
No, a CFS is not required in this case.
An “action task” is a task of the project described in Annex 1.
For information on tax regulations in a specific country, please contact your internal support facilities or tax consultant.
Please follow the EU regulations on IVD: http://www.namsa.com/blog/View/entryid/190/blogid/2/update-on-the-new-eu...
Yes it is necessary. The ethical approval will only be granted if the CT will be performed according to GCP.
Yes.
You should mention the national Ethic regulations of the respective countries and state that you will perform your trials in accordance to those.
Please have a look at the FFH website: http://www.fitforhealth.eu/news/new-fit-health-20-partner-search-and-mat....
That is what EC told us. Sorry we do not know more on that.
It is possible to include a CRO.
There is a partner search tool on the FFH2.0 website which you may want to check out for this purpose: http://mm.fitforhealth.eu
Yes, costs of insurance can be part of the unit costs.
Depends on the topic. They also fund observational studies, if there is a respective topic for that.
Yes that is correct. Each beneficiary can decide to be reimbursed on basis of unit costs or actual costs for a given clinical study.
100%
Any costs that will be reimbursed by health care providers/ insurance are NOT eligible for funding via the EU project.
Unit costs are partner specific - i.e. you will most likely have different unit costs for each partner.
Yes - they would have to be defined for that linked third party specifically in any case.
Yes, but not in a separate template.
Yes, and as topic says: you should have granted the orphan drug designation at the time of stage 2 submission.
The topic says “based on clinical trials and/ or real world data”. Duration depends fully on your concept and approach.
I understand the topic PHC 12 in a way that it can be and/ or medical device, so I would say, the evaluators have decided accordingly.
In that case you have only one reporting period.
That depends on the topic.
No, project funded in 2019 for example will definitely continue after 2020.
There are no rules on the duration of H2020 projects. It depends on the nature of your project. The average project duration is between 3-5 years.
Say as it is – we have this, we are here, but xyz is not yet precisely projectable.
Not particularly.
As a separate document.
Yes, this was a technical mistake.
There are certainly other funding opportunities as well. Fit for Health is specialized in providing support for Horizon2020 participation though.
Societal Challenge.
To be eligible as a coordinator , or getting financial support in some conditions, a company needs to have its financial viability checked The Commission provides this user-friendly electronic tool for applicants so that they could simulate the financial viability check of their organisation for their own information. The tool uses the ratios described in the "Rules on verification of existence, legal status, operational and financial capacity". It is available on "Partcipant Portal" at http://ec.europa.eu/research/participants/portal/desktop/en/organisations/lfv.html
Nike
As you posted your question in Fit for Health website you might be particularly interested in the rules in the Health topic in which the US have a special status. Due to a bilateral agreement US partners are having automated access to funding from H2020. It is important to understand that this only applies to the topic 'Health'.
You can find this in the footnote 29 on page 73 of the Health Work Programme: 'In recognition of the opening of the US National Institutes of Health’s programmes to European researchers, any legal entity established in the United States of America is eligible to receive Union funding to support its participation in projects supported under all topics in calls under the Societal Challenge ‘Health, demographic change and well-being’.
Only exception is PHC12 (SME Instrument) in which appicants have to be established in a MS or AC (whether subcontracting to a US partner is possible is not specified so I would contact the programme officers about this.)
For all other topics in H2020 , If a grant applicant can convincingly demonstrate that without the participation of a US partner the objectives or impact of the proposal cannot be achieved, the EC can allow the US partner to access funding. But the case has to be really convincing. If you have a US partner who has a specific expertise, a patent, market access or anything that cannot be found within the EU then you may have such a case. What you should definitely avoid is to make the impression that you look for EU funding for research activities and that a significant part of the exploitation or market uptake (hence a large part of the expected turnover) will be generated in the US.
However, if the US partner for example allows a European SME to access the US market, this would be a pro for adding a US partner.
In any case, if you are planning to involve a US partner we strongly recommend to contact the assigned project officer or at least your NCP to get a better idea about the official take on things
You can find a list hereunder, describing the various instruments in Horizon 2020 and the according eligibility conditions
| Eligibility conditions | |
|---|---|
| Research & innovation action | Three legal entities. Each of the three shall be established in a different Member State or associated country. All three legal entities shall be independent of each other. |
| Innovation action | Three legal entities. Each of the three shall be established in a different Member State or associated country. All three legal entities shall be independent of each other |
| Coordination & support action | One legal entity established in a Member State or associated country. |
| SME instrument |
One for-profit SME. Only applications from SMEs established in EU Member States or countries associated to Horizon 2020; No concurrent submission or implementation with another phase 1 or phase 2 project. |
|
ERA-NET Cofund |
Three legal entities. Each of the three shall be established in a different Member State or associated country. All three legal entities shall be independent of each other. Participants in ERA-NET Cofund actions must be research funders: legal entities owning or managing public research and innovation programmes |
| Pre-commercial procurement (PCP) Cofund & Public procurement of Innovative solutions (PPI) Cofund |
Three legal entities. Each of the three shall be established in a different Member State or associated country. All three legal entities shall be independent of each other. A minimum of two independent legal entities which are public procurers from two different Member States or associated countries. |
Nike Roshe Run Men
You can find a series of health-related topics in doing a keyword search under the European Commission > Research & Innovation > Participant portal
Click Here to search for topics by keywords
In addition, see all the open calls of Marie Sklodowska-Curie actions (MSCA) and European Research Council (ERC) grants for individual researchers of any nationality and research teams. The MSCA grants include opportunities for companies and SMEs. The MCSA and ERC grands are not restricted to specific topics.
adidas superstar damen glitzer silberWhat means Horizon 2020 and what will be the main advantages for me as an SME?
You can find a detailed description about the participation in the new SME-instrument, in the collaborative projects, as well as information about access to Debt and Equity Financing in the Participants portal.
NikeAn application for a collaborative project is not interfering with an application for the SME instrument.
Yes, the restriction is only WITHIN the SME instrument
As subcontractor that MIGHT be possible, but as PARTNER definitely NOT
The high growth potential is intended to help Europe achieve the EUROPE 2020 goals which is: jobs, jobs, jobs!
A complementary funding is not excluded in the work programme.
The 50.000 EUR can be used for any kind of activity (personnel costs, subcontracting, costs for patent search etc).
Yes for the PHC topic, funding rate is 100%
Please use the definitions as shown here (and please see also the corresponding literature)
A biomarker is a characteristic that is objectively measured and evaluated as an indicator of normal biologic processes, pathogenic processes, or pharmacologic responses to a therapeutic intervention (NHI Biomarkers Definitions Working Group; 2001)
A valid biomarker is defined as “a biomarker that is measured in an analytical test system with well-established performance characteristics and for which there is an established scientific framework or body of evidence that elucidates the physiologic, toxicological, pharmacologic, or clinical significance of the test results
(FDA. Guidance for industry - pharmacogenomic data submissions. 2005)
Air Force 1They will be rejected with the possibility to resubmit an improved version.
Yes. You can download it under http://ec.europa.eu/research/participants/data/ref/h2020/call_ptef/pt/h2020-call-pt-sme-1_en.pdf
Accesorios para el runningAny SME must be (re)checked, if it was not validated within the last two years.
At least 2 evaluators are planned to evaluate a proposal.
Yes, reporting periods such as once a year; the general rules H2020 apply.
EASME is working on a database that is not ready yet. EEN will be included in this.
The SME instrument is targeted at all types of innovative SMEs showing a strong ambition to develop, grow and internationalise. It provides staged support covering the whole innovation cycle in three phases complemented by a mentoring and coaching service. Transition from one phase to the next will be seamless provided the SME project proves to be worth further support in a further evaluation. Each phase is open to new entrants.
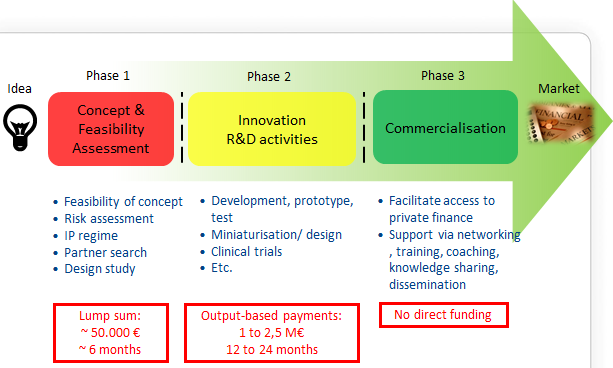
Description: Feasibility study verifying the technological/practical as well as economic viability of an innovation idea/concept with considerable novelty to the industry sector in which it is presented (new products, processes, design, services and technologies or new market applications of existing technologies). The activities could, for example, comprise risk assessment, market study, user involvement, Intellectual Property management, innovation strategy development, partner search, feasibility of concept and the like to establish a solid high-potential innovation project aligned to the enterprise strategy and with a European dimension. Bottlenecks in the ability to increase profitability of the enterprise through innovation shall be detected and analysed during phase 1 and addressed during phase 2 to increase the return in investment in innovation activities.
Funding rate: Funding will be provided in the form of a lump sum of EUR 50,000 (70% funding rate).
Description: innovation projects that address a specific challenge and demonstrate high potential in terms of company competitiveness and growth underpinned by a strategic business plan. Activities should focus on innovation activities such as demonstration, testing, prototyping, piloting, scaling-up, miniaturisation, design, market replication and the like aiming to bring an innovation idea (product, process, service etc.) to industrial readiness and maturity for market introduction, but may also include some research. SMEs can subcontract or buy in work and knowledge that is essential for their innovation project in the spirit of the innovation voucher concept. Proposals should be based on a strategic business plan either developed through phase 1 or another means.
Funding rate: 70% [or in exceptional cases, where the research component is predominant, the reimbursement rate may be 100%, as specified in each topic].
Support to commercialisation promotes the wider implementation of innovative solutions and customers and supports financing of growth by facilitating access to public and private risk capital. This stage will not provide for direct funding, but SMEs can benefit from indirect support measures and services as well as access to the financial facilities supported under Horizon 2020.
Each beneficiary of the SME instrument will be offered business coaching support during Phase 1 (up to 3 coaching days) and Phase 2 (up to 12 coaching days) in addition to the grant offered. This support will be provided through the Enterprise Europe Network (EEN) and delivered by a group of qualified and experienced business coaches. The local EEN office will introduce the beneficiary to the coaching process and propose a selection of coaches from the database managed by the Commission for the beneficiary to choose from. The objective is to accelerate the impact of the support provided through the SME instrument and to equip beneficiaries with the necessary skills, business processes and relevant competencies for long-term growth. Phase 3 does not include individual business coaching, but SME instrument participants will be able to count on continuing EEN support in linking to relevant support services within the Network, regionally or nationally. It is important to note that the objective of coaching is not to support the company in project management or reporting obligations related to Horizon 2020 participation.
ADIDASPlease use the Fit for Health 2.0 support network and publish a profile on the Fit for Health 2.0 match-making facility
NikeThe type of protection needed depends on the nature of the results. In case Intellectual Property Rights come out as necessary it should be recommended choosing the adequately IPR that suit with the aim of the project and be the most cost efficiently. Grant agreement contents specific rules to follow, for instance, the obligation to adequately and effectively protect the results got (“foreground”) and that is capable to industrial and commercial replication.
Air Max 90 YEEZY 2 SPIn principle yes.
Yes, subcontracting of clinical studies is possible.
Clinical trials can be subcontracted.
Out-licensing after Phase 2 can be considered as market uptake.
There is no exclusion of medical devices.
That is a risk you should address in your application.
Clinical trials can be sponsored. But the rules of this instrument have to be considered.
No, it is not possible.
Yes, this is allowed.
Fit for Health 2.0 has a large database with companies and organisations interested in the health area of Horizon 2020.
As far as they comply with the FTI goals (delivery of a new educational concept in three years), then it is possible.
Not as a partner.
Yes, there is no exclusion of small markets.
Universities are also eligible and they are reimbursed at a 100% rate.
They can apply.
In the case this new application addresses a challenge under horizon 2020 and is innovative enough yes.
Yes.
Then it is not research infrastructure, but deployment infrastructure that should be checked in your specific case by your health NCP and SME-NCP.
Unfortunately not. Actions should focus on bringing a product or service to the market.
It is surely a decision criterium with equal points among several applications.
Yes, this is right.
Unfortunately, some of those documents are not available.
Reviewers are briefed by the agency taking care of the evaluation (EASME).
Yes, as far as these processes are needed for an innovative service or product to be pushed to the previous steps before market uptake.
No. FTI is a grant.
Only development of most innovative products or services will be funded. If these are needed for scale up, then yes.
There is no explicit limit for subcontracting.
This is not possible.
You must already provide a rough strategy in your application. Design of tailored strategies can be funded.
Yes, as well as the preparation and fill of a patent.
If you mean analytical services, yes.
As in any other Horizon 2020 project: personal, consumables, subcontractors...
Research should not be a main focus of an FTI action, so equipment for research is difficult to get funded.
For all innovation activities, a flat rate for indirect costs applies.
“on the market” can be sale of device after completion of clinical trials. But out-licensing after Phase 2 can be considered as market uptake.
There is not a rule for that.
You should have the EU market in focus. If you can also deliver solutions for additional markets you can do so.
You should have the EU market in focus. If you can also deliver solutions for additional markets you can do so.
Yes, 36 months from project start.
A dissemination plan is highly dependent on your project! There is not such a template.
There are no limitations for target markets.
There is no penalty. You must convincingly explain this time to market, which risks you could face and how you could overcome them.
No, there is no limitation.
The PIC number must be new and not been used for FP7 projects
You must sound convincing in your application and offer solutions for overcoming that risk
This is right.
No, you must be a first-time applicant, meaning that you receive your PIC now for the first time.
You must convincingly explain in your application in which TRL step your product/service is.
It depends on the country. In Germany SME-NCPs work together with the thematic NCPs.
You can find a comprehensive list under: http://ec.europa.eu/research/participants/portal/desktop/en/support/nati...
PIC number is related to your organisation. Your organisation should register for a PIC number.
The IMI Project COMBACTE deals with antimicrobial resistance (AMR) http://www.imi.europa.eu/content/combacte
In Germany only for stage 2, not for the EoI preparation
I suggest to check the IMI Newmeds project as an example: http://www.imi.europa.eu/content/newmeds
Call 9 has unfortunately already closed, but clinical trials can be funded within IMI projects
When patient data are in use, research on animals, stem cells, and others specified on cordis web pages in section ethical issues…
Two options:
You use the local and actual salary rates. You can use Lump Sums for International Cooperation Partner Countries (ICPC) ICPC got the option being reimbursed: the basis of eligible costs or may opt for lump sums lump sum contribution is defined per country income group in the „list of ICPC economies“:
Economy contribution (€/researcher/year) low income 8 000 €
lower middle income 9 800 €
upper middle income 20 700 €
upper funding limits have to be applied disadvantage of lump sum payment: it is deemed to cover all costs (see also: Guide to Financial Issues, p. 69 ff)
Zapatillas Running BaratasThis depends on the type of project and kind of organization ( SMEs, Universities, and/or others…) Please refer to the guidelines and Participant portal
NikeThe key question is if the cost is a subcontract. If the services in question are a subcontract then the subcontract can not be taken into account when calculating overheads. The classification of the services depends on their character.
Some costs incurred in relation to organisation of the meeting may be considered as subcontracting (e.g. catering services provided by an external company) whereas others (renting the rooms directly in a hotel) would not fall within this category. ln this sense remember that subcontracting is a business transaction by which the subcontractor performs some work for a beneficiary.
Subcontracting costs are direct costs. Whether major or minor costs, they have to be identified by beneficiaries in the financial statement form (Form C, Annex VI to GA). In any case, they should be reported as subcontracting (if you are paying for a service; the difference is that the GA allows that these minor subcontracts do not previously appear in the Description of work of the project, As subcontracts, they are a cost to a beneficiary for a work/service which is performed by a third party and not by the beneficiary, and therefore indirect costs can not be charged by the beneficiary on them; in this cases, the indirect costs are already covered by the price paid by the beneficiary to the subcontractor. The same rules for subcontracting apply to all projects, including CSA.
Gifts for RunnersThe 375k€ limit applies for each project separately.
In our project we need to follow the progress of patients from different wards, GPs, home nursing organisations. They need to all fill-out a questionnaire and need to have their costs covered for this. Classical sub-contracting. There will be many (up to hundreds) of different organisations. We can't have a contract with them exactly, nor can we write out a call for the sub-contracting. What do we need to watch out for to ensure we follow the EU rules for sub-contracting?
In this case there should be a detailed description of the necessity of these people working in the project under Subcontracting in Annex I GA /DoW – this should be discussed with the PO/FO in detail as well as the costs.
FootwearYes, of course. You are the owner.
Daily allowances are accepted by the commission if they are part of your normal operating accounting principles (“usual practice”).
For clarity: dissemination includes everything from attending conferences to talk about results of the project to personnel costs for writing scientific papers about the project results to establishing and maintaining the project website. Is this correct?
Dissemination is everything you do to get the results / outcome of the project to the right target group(s)Air Max 90 Ultra 2.0 Essenti
The advice is to include an AB already in the project governance structure of the project at the first stage. It may help to convince the evaluators.
Our centre is a national consortium composed of research groups from different hospitals, universities, etc. If a research group applies for funds as part of the consortium, how do we reflect their belonging to other institutions?
Here it depends if your centre is a legal entity or not. If it is, then the Centre can apply for funding. If not, the hospitals, universities etc. should apply as partners. If the centre is a partner and people doing the research work are not “directly hired” by the centre but have a working contract at a hospital, university which is member of the centre, then the costs of the personnel might only be eligible if the hospital, university is as a “third party linked to the beneficiary” with special clause 10 in the Grant Agreement working in the project.Nike Ambassador IX 9
Can you please tell me what the situation is with reporting in FP7? One of our deliverables should already be completed by Month 5 which has just passed. I have the information for the report. Should this be uploaded and submitted online now or can we wait for the reports of the 1st reporting period, after 18 months?
If you have arranged Month 5 for this deliverable in your Annex 1, this should be uploaded now. Here are some references from the relevant documents:
Guidance notes for reporting: 2.1 During the course of the project, to be submitted:
1. The deliverables identified in Annex I to the Grant Agreement, according to the timetable specified in the Deliverables list.
Guide for Applicants Health Theme: Please note that each deliverable will have to be submitted as a distinct document/report. In order to keep your deliverables manageable, small related deliverables should be grouped as specified parts (equivalent to 'subdeliverables') of a single more substantial deliverable. Progress towards achievement of the full deliverable can then be demonstrated in the periodic reports by reference to the smaller parts. The full deliverable will only be submitted when all parts have been -completed. Ideally this will be at the same date as a periodic report.
Jordan Hydro 6 SandalsMy question is in relation to direct and indirect cost in FP7. If we allocate some of the time of the Director of the Research Centre, where do we allocate this cost? As direct costs or personnel costs?
Here it depends if the Director receives a salary or not. If the Director does not receive any salary the Marie Curie rates can possibly be taken. Otherwise there has to be made the distinction if his salary is completely in the overhead costs (indirect costs). If so and he wants to get reimbursed hours in the project, these hours have to be taken out of the indirect costs and booked under the direct costs – only then these costs can be reimbursed under personnel costs. I just add here that a complete time recording for the Director is needed too.
Supreme X Air Max 98faq
Yes, it is forbidden. Only complementary financing is allowed.
FFH 2.0 experts offer advice and guidance for SMEs throughout all phases of the innovation cycle
Austrian Research Promotion Agency